Аннотация
Роль микробиома кишечника в здоровье животных становится все более очевидной. Хотя структура микробиоты кишечника A. mellifera хорошо известна, мало что известно о динамических изменениях на разных стадиях развития. В данном исследовании мы изучили динамические изменения микробиоты кишечника A. mellifera на разных стадиях развития, охватывающих весь жизненный цикл, с помощью высокопроизводительного секвенирования генов 16S рРНК. Результаты показали, что основная (общая) микробиота кишечника значительно изменяется на разных стадиях развития. Разнообразие бактериального сообщества у рабочих разного возраста значительно отличалось. Кроме того, сравнивая основную микробиоту кишечника у рабочих разного возраста, мы обнаружили, что у недавно появившихся рабочих микробиота была меньше. Три рода, Gilliamella, Frischella и Snodgrassella, были значительно колонизированы в 1 день после слияния (dpe); Lactobacillus, Bifidobacterium, Commensalibacter были значительно колонизированы в 3 дня после слияния и значительно уменьшились у Gilliamella. Lactobacillus kunkeei и Bartonella были значительно колонизированы на 12 дпе и были значительно снижены Lactobacillus helsingborgensis. Commensalibacter и Bifidobacterium были значительно снижены на 25 дпе, а Bacteroides, Escherichia-Shigella и Porphyromonadaceae были значительно снижены между 19 и 25 дпе. Наши результаты показывают смену микробиоты кишечника рабочих от рождения до старения, что обеспечивает теоретическую основу для дальнейшего изучения роли микробиоты кишечника на разных стадиях развития.
1.Введение
Медоносная пчела, A. mellifera, является важным экономическим насекомым. По оценкам правительства США, ежегодная социальная прибыль медоносных пчел составляет от 1,6 до 5,7 миллиардов долларов (Southwick and Southwick, 1992). Медоносные пчелы также являются важными опылителями; с 2006 по 2008 год среднегодовая стоимость опыления пчелами достигла 304,22 млрд юаней, что эквивалентно 12% от общей стоимости сельскохозяйственной продукции Китая (Liu et al., 2011). Однако в последние годы выживание медоносных пчел оказалось под большой угрозой. В частности, в США было зарегистрировано расстройство распада колоний (CCD), при котором тысячи ульев оказались пустыми с таинственным исчезновением пчел (Stokstad, 2007). Это происшествие вызвано многими факторами, включая изменение климата (Langowska et al., 2017), интенсивное использование пестицидов (Colin et al., 2019), злоупотребление антибиотиками (Owen, 2017) и заражение патогенами (Cameron et al., 2011). Все эти факторы влияют на кормление и другие виды поведения медоносных пчел. Микробиота кишечника медоносных пчел имеет тесную связь с хозяином и обеспечивает ему ряд преимуществ, таких как содействие перевариванию пищи, необходимых питательных веществ, деградация токсичных компонентов, защита от патогенов, а также регуляция развития, поведения и иммунитета хозяина (Moran et al., 2003
Согласно анализу 16S рДНК сообщества в кишечнике, исследователи обнаружили, что в кишечнике рабочих особей распространены в основном девять видов бактерий (Martinson et al., 2011; Sabree et al., 2012), пять из которых присутствуют почти у всех медоносных пчел, включая два грамотрицательных вида, Snodgrassella alvi и Gilliamella apicola (Kwong and Moran, 2013). Среди этих бактерий Lactobacillus firm-4 и firm-5, относящиеся к филуму firmicutes, были наиболее доминирующими и широко распространенными в кишечнике (Babendreier et al., 2007). Численность Bifidobacterium относительно ниже по сравнению с большинством видов бактерий, но они широко распространены (Bottacini et al., 2012a). По сравнению с основными бактериями, численность и распространение Frischella perrara (Philipp et al., 2013), Bartonella apis (Jeyaprakash et al., 2003), Parasaccharibacter apium (Vanessa et al., 2014) и «Alpha 2.1» не являются стабильными. Медоносные пчелы являются хорошей моделью для исследования микробиоты кишечника. Андерсон и др. (2018) изучали рабочих и маток с разными возрастными фенотипами и обнаружили, что, несмотря на то, что рабочие и матки имеют много общих видов кишечных бактерий, структура микробиоты кишечника между ними заметно отличается (Anderson et al., 2018). Их исследование также показало, что накопление углерода у королев значительно коррелирует с увеличением количества Lactobacillus и Bifidobacterium и уменьшением филума Proteobacteria (Anderson et al., 2018). Кроме того, косвенно доказано, что микрофлора кишечника пчел также играет важную роль в борьбе с патогенными микроорганизмами. Предыдущие исследования показали, что численность этих основных бактерий кишечника была относительно ниже, чем Crithidia bombi у шмелей, а пересадка фекалий вновь развившихся взрослых шмелей дала аналогичный эффект (Hauke and Paul, 2011). Однако, когда структура микрофлоры кишечника нарушается антибиотиками, разнообразие микрофлоры кишечника уменьшается, и медоносные пчелы с аномальными бактериями кишечника будут более восприимчивы к оппортунистическим патогенам, что снижает выживаемость (Raymann et al., 2017). Аналогичные исследования были проведены на Apis cerana путем изучения A. cerana, зараженной Nosema ceranae. Структура и разнообразие микрофлоры кишечника были значительно изменены, что привело к увеличению смертности (Huang et al., 2018). Эти результаты демонстрируют важность бактерий кишечника медоносной пчелы для здоровья хозяина.
Развитие технологии высокопроизводительного секвенирования значительно продвинуло исследования микробиоты кишечника (Ross, 2012). Более четкое понимание состава микрофлоры кишечника пчел постепенно формировалось с помощью методов независимого культивирования, в частности высокопроизводительного секвенирования гена 16S рРНК (Kwong and Moran, 2016). Например, Джонс и др. секвенировали геном 16S рРНК рабочих, подвергавшихся воздействию различных факторов окружающей среды, и обнаружили, что воздействие различных факторов окружающей среды влияет на относительное обилие частично микробных групп (Jones et al., 2017). Какуману и др. обнаружили, что воздействие пестицидов значительно повлияло на структуру микрофлоры кишечника у рабочих медоносных пчел, используя технологию высокопроизводительного секвенирования (Kakumanu et al., 2016). Далее Мотта и др. изучили влияние глифосата на микрофлору кишечника пчел и обнаружили, что воздействие может нарушать жизнедеятельность полезных кишечных бактерий (Motta et al., 2018).
Несмотря на множество предыдущих исследований, посвященных микробиоте кишечника A. mellifera, динамическая смена микрофлоры кишечника A. mellifera на протяжении всего жизненного цикла изучена слабо. Здесь технология высокопроизводительного секвенирования была использована для анализа микробиоты кишечника рабочих особей на 0 день после всхода (dpe), 1 dpe, 3 dpe, 7 dpe, 12 dpe, 19 dpe, 25 dpe, 30 dpe, 35 dpe и 40 dpe. Были освещены динамические изменения в микробиоте кишечника рабочих, с целью изучения последовательности развития рабочих от рождения до старения и дальнейшего понимания динамических изменений в сообществе микробиоты кишечника рабочих. Эти результаты представляют собой ценный генетический ресурс для использования микробиоты кишечника с целью улучшения состояния здоровья пчел в будущем.
2.Материалы и методы
2.1. Отбор проб рабочих пчел
Мы собирали рабочих пчел (Apis mellifera) в городе Куньмин, провинция Юньнань, Китай, с начала июля по конец августа 2018 года. Для подтверждения возраста рабочих мы выбрали рамку из группы здоровых колоний, удалили из нее рабочих и поместили их при температуре 34 °C в темноте (имитируя условия в улье). Сначала мы случайным образом отобрали 6 рабочих, которые только что вышли из коконов в возрасте 0 dpe, затем сразу же поместили их в центрифужную пробирку объемом 1,5 мл и хранили в ультранизкотемпературном морозильнике (EU1DW/BD-55W321EU1, Китай) при -80 °C (образцы не контактировали с пчелами-воспитательницами). После этого рамки возвращали в темную инкубационную среду. Затем 300 вновь появившихся рабочих были помечены эмалевой краской Testors и помещены обратно в исходный улей для естественного роста. Шесть рабочих были случайным образом собраны на 1 дпе, 3 дпе, 7 дпе, 12 дпе, 19 дпе, 25 дпе, 30 дпе, 35 дпе и 40 дпе. После вскрытия кишечника в стерильной среде весь кишечник был удален и помещен в центрифужную пробирку объемом 1,5 мл. Все образцы хранились в 75% спирте в ультранизкотемпературной морозильной камере при — 80 °C для выделения ДНК.
2.2. Выделение ДНК и ПЦР-амплификация
Далее ДНК микробиоты кишечника была выделена с помощью набора E.Z.N.A.® soil DNA Kit (Omega, США) в соответствии с протоколами производителя. Конечную концентрацию и чистоту ДНК определяли с помощью УФ-vis спектрофотометра NanoDrop 2000 (Thermo Scientific, США), а качество ДНК проверяли электрофорезом в 1% агарозном геле. Гипервариабельные области V3-V4 бактериального гена 16S рРНК амплифицировали с праймерами 338 F (5′- ACTCCTACGGGAGGCAGCAG-3′) и 806R (5′-GGACTACHVGGGTWTCTAAT-3′) с помощью ПЦР-системы термоциклера (Applied Biosystems, США). ПЦР проводили по следующей программе: 3 мин денатурации при 95 °C, 27 циклов по 30 с при 95 °C, 30 с для отжига при 55 °C и 45 с для элонгации при 72 °C, и окончательное удлинение при 72 °C в течение 10 мин. ПЦР проводили в трех экземплярах в 20 мкл смеси, содержащей 4 мкл 5 × FastPfu Buffer, 2 мкл 2,5 мМ dNTPs, 0,8 мкл каждого праймера (5 мкМ), 0,4 мкл FastPfu Polymerase и 10 нг шаблонной ДНК. Продукты ПЦР очищали с помощью набора AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, США) и затем количественно определяли с помощью наборов QuantiFluor™-ST (Promega, США) в соответствии с протоколом производителя.
2.3. Секвенирование на Illumina MiSeq и обработка данных секвенирования
Очищенные ампликоны объединяли в эквимолярные пулы и проводили попарное секвенирование (2 × 300 п.н.) на платформе Illumina MiSeq (Illumina, США) в соответствии со стандартными протоколами Majorbio Bio-Pharm Technology Co. Ltd. (Шанхай, Китай).
Необработанные файлы fastq были отфильтрованы с помощью программы Trimmomatic (версия 0.36) и объединены с помощью программы FLASH (версия 1.2.7) (Zhang et al., 2018) с учетом следующих критериев: (i) Чтения были усечены на любом участке, получившем средний балл качества <20 в скользящем окне 50 п.н. (ii) Последовательности с перекрытиями длиной более 10 п.н. объединялись в соответствии с их перекрытием с несовпадением не более 2 п.н. (iii) Последовательности каждого образца разделялись в соответствии со штрихкодами (точное совпадение) и праймерами (допускается несовпадение 2 нуклеотидов), и чтения, содержащие неоднозначные основания, удалялись. Затем была проведена кластеризация операционных таксономических единиц (OTU) на последовательности со следующими критериями: (i) Из оптимизированных последовательностей были извлечены неповторяющиеся последовательности для сокращения избыточных вычислений в процессе анализа (http://drive5.com/usearch/manual/dereplication.html). (ii) Одиночные последовательности, которые не повторялись, были удалены (http://drive5.com/usearch/manual/singletons.html). (iii) Операционные таксономические единицы (OTU) были кластеризованы с отсечением 97% сходства с помощью UPARSE (версия 7.1 http://drive5.com/uparse/) с новым «жадным» алгоритмом, который выполнял фильтрацию химер и кластеризацию OTU одновременно. Таксономию каждой последовательности гена 16S рРНК анализировали с помощью алгоритма RDP Classifier (http://rdp.cme.msu.edu/) по базе данных SILVA (SSU123) 16S рРНК с использованием порога достоверности 70%. Кроме того, был проведен анализ разреженности с помощью программы Mothur.
2.4. Статистический анализ и сравнение микробных сообществ
Тест Kruskal-Wallis был использован для выявления наличия существенных различий в альфа-метрике разнообразия между десятью группами (ACE, Chao1, Shannon и Simpson). Согласно полученным данным о численности сообществ, статистический метод Kruskal-Wallis был использован для выявления различий относительного богатства OTUs в микробных сообществах разных групп, а для оценки значимости (P-значения) можно провести гипотетический тест. Кроме того, была проведена коррекция FDR для P-значения множественных тестов, а для сравнения двух групп использовался статистический U-тест Манна-Уитни. В качестве программного обеспечения использовался пакет stats в R. Результаты с P < 0,05 между группами были признаны статистически значимыми.
3.Результаты
3.1. Сводка данных секвенирования
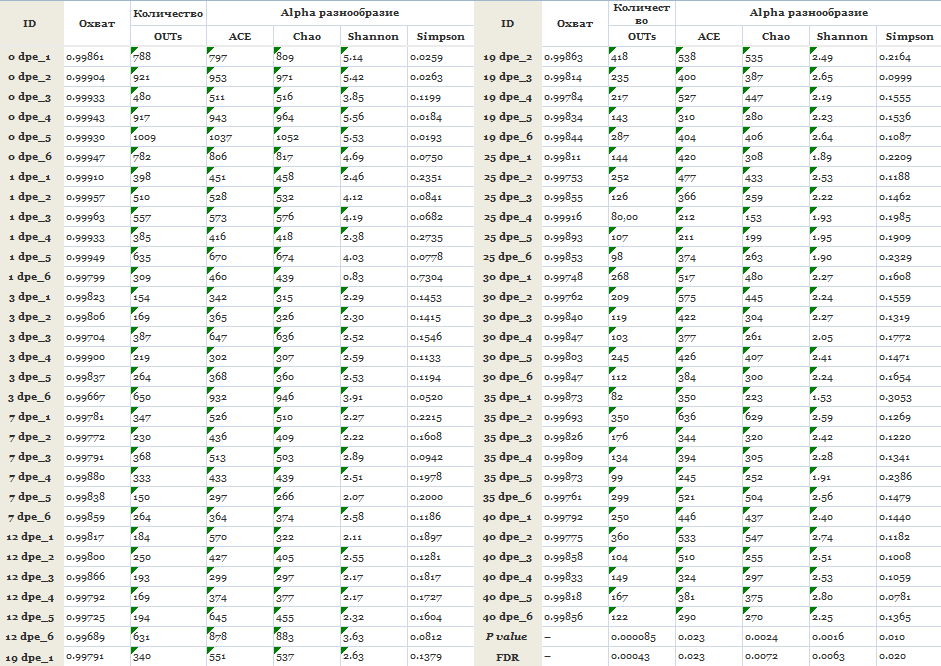
В общей сложности 3 204 891 высококачественная последовательность была получена из 60 рабочих образцов, средняя длина последовательности составила 444 п.н.. Среди последовательностей было выявлено 3217 бактериальных операционных таксономических единиц (OTU) при 97% сходстве последовательностей. Мы оценили OTU% в 60 образцах медоносной пчелы по индексу охвата Гуда, и средний бактериальный охват составил 0,99 ± 0,00069. Это указывает на то, что полученные данные могут адекватно охватить микрофлору кишечника медоносных пчел. Кроме того, кривые разрежения имели тенденцию приближаться к плато насыщения, что указывало на то, что объем данных секвенирования был разумным; большее количество данных позволило бы получить лишь небольшое число новых OTU. Кроме того, кривые разрежения также показали, что богатство сообщества в 0 дпе было самым высоким среди всех временных точек отбора проб (Дополнительный рис. S1).
3.2. Сравнение индексов разнообразия бактериального сообщества
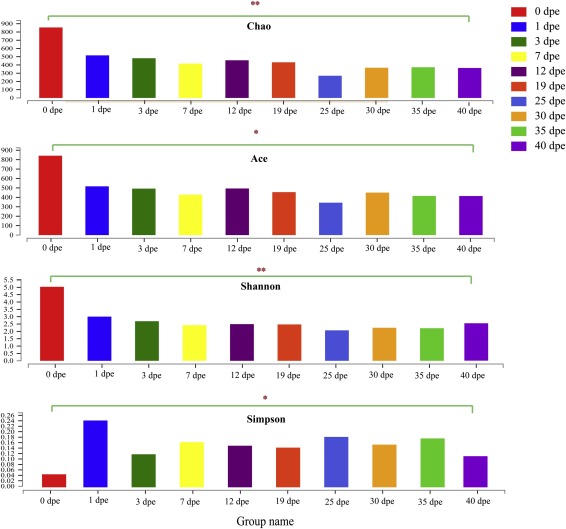
Богатство сообщества оценивали по индексам ACE и Chao1, а разнообразие сообщества определяли по индексам Шеннона и Симпсона для десяти групп. С помощью теста Kruskal-Wallis для сравнения мы обнаружили, что индексы Шеннона, ACE и Chao статистически значимо отличались между разновозрастными рабочими (Таблица 1). Этот результат показал, что разнообразие и обилие микрофлоры кишечника зависело от возраста рабочих (рис. 1). P-значение и FDR значимости индекса разнообразия были следующими: ACE, P = 0,023, FDR = 0,023; Chao, P = 0,0024, FDR = 0,0072; Shannon, P = 0,0016, FDR = 0,0063; и Simpson, P = 0,010, FDR = 0,020. Кроме того, были обнаружены значительные различия в количестве микробных OTU среди разновозрастных рабочих (Number of OTUs, P = 0,000085, FDR = 0,00043), что указывает на то, что изменение количества OTU значительно отличалось между стадиями возраста хозяина (Таблица 1).
3.3. Динамика микробной флоры у разновозрастных работников
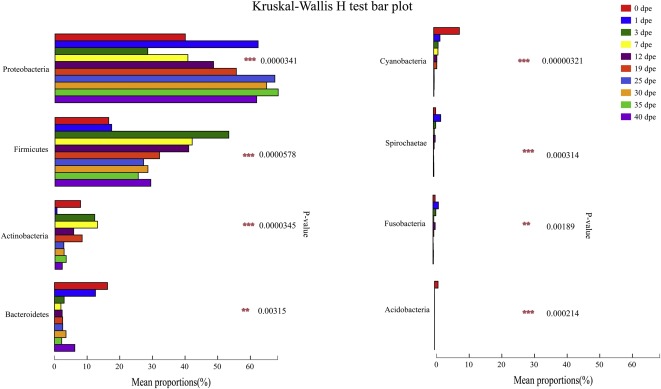
Все последовательности, полученные в данном исследовании, были классифицированы на уровне филумов и родов. Всего было идентифицировано 41 фила и 838 родов (рис. 2 и рис. 3). Репрезентативные последовательности на уровне филумов представлены на рис. 2. Филы Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, Cyanobacteria, Spirochaetae, Fusobacteria, Deinococcus-Thermus, Verrucomicrobia, Chloroflexi, Acidobacteria и Planctomycetes были доминирующими филами, что аналогично предыдущим исследованиям (Yun et al., 2018). На относительное обилие фил в кишечной флоре существенно влияли разновозрастные стадии рабочих. У недавно появившихся рабочих доминирующими филами были Proteobacteria, Firmicutes, Bacteroidetes, Cyanobacteria и Acidobacteria, а относительное обилие Proteobacteria и Firmicutes значительно увеличивалось по мере развития рабочих (рис. 4). P-значение и значения FDR были определены путем межгруппового сравнения и были следующими: Proteobacteria (p = 0,0000341, FDR = 0,000129), Firmicutes (p = 0,0000578, FDR = 0,000171), Actinobacteria (p = 0,0000345, FDR = 0,000129), Bacteroidetes (p = 0,00315, FDR = 0. 00561), Cyanobacteria (p = 0.00000321, FDR = 0.0000284), Spirochaetae (p = 0.000314, FDR = 0.000715), Fusobacteria (p = 0.00189, FDR = 0.00352) и Acidobacteria (p = 0.000214, FDR = 0.000517).
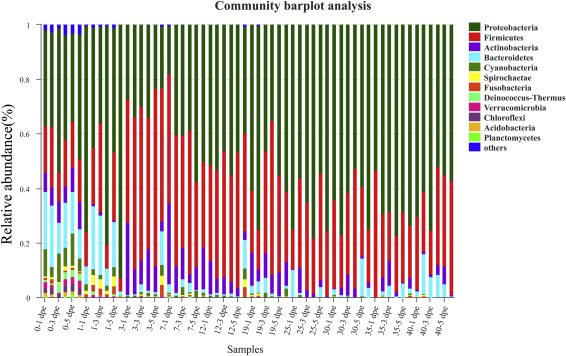
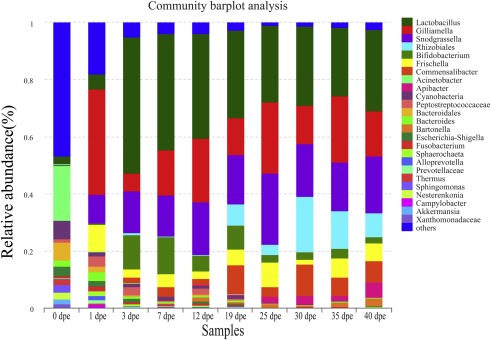
Пчелы в возрасте 0 дпе демонстрировали самое высокое бактериальное разнообразие, среди которых Bacteroidetes (15,82%), Cyanobacteria (7,79%) и Verrucomicrobia (1,94%) были доминирующими филами по сравнению с другими однодневными рабочими (Дополнительный рис. S2). На 1 дпе Bacteroidetes снизились до 12,37%; на 3 дпе Bacteroidetes имели тенденцию к стабилизации. До 19 дпе во всех группах наблюдалось сходное распределение состава кишечной флоры на уровне филумов, однако относительное обилие доминирующих бактерий менялось по мере роста рабочих. На 25 дпе было обнаружено увеличение разнообразия микрофлоры кишечника на уровне филумов, но состав был очень похож на тот, что был до 19 дпе.
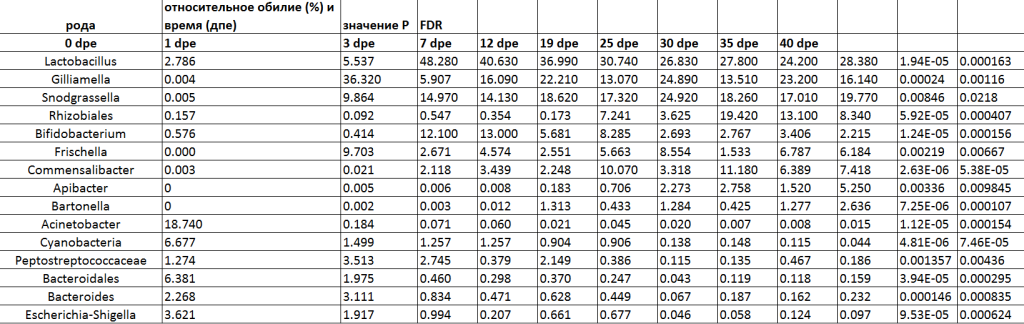
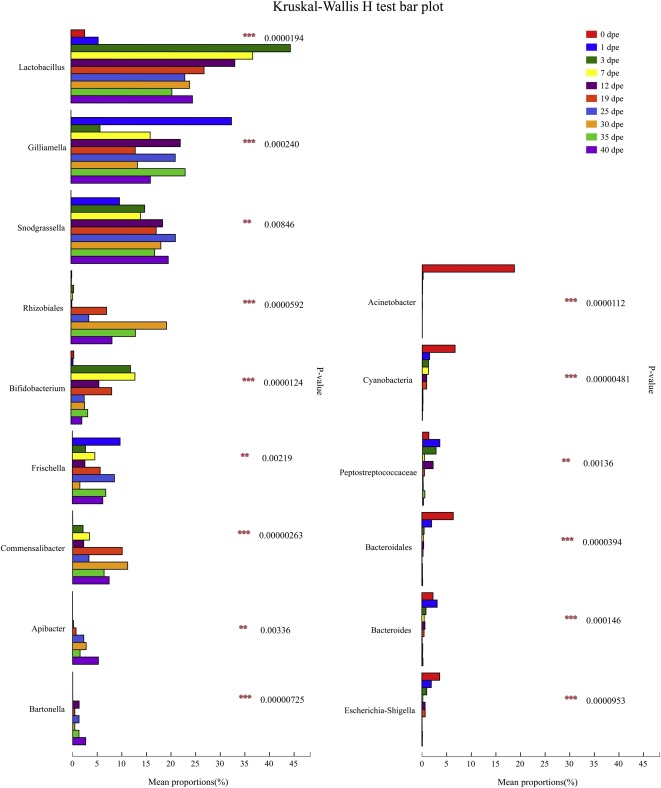
На 0 дпе микробиота кишечника рабочих состояла в основном из Acinetobacter, Bacteroides и Cyanobacteria, а на 1 дпе Acinetobacter был заменен другими бактериями. Эти три филы оставались в микробиоте кишечника рабочих до 19 дпе, а на 25 дпе их относительная численность составляла менее 0,9%. Кроме того, стоит отметить, что на 0 дпе в составе микрофлоры кишечника рабочих отсутствовали пять основных бактерий (A. mellifera). Мы обнаружили, что численность основных бактерий в микробиоте разновозрастных рабочих существенно различалась (табл. 2). Относительная численность рода Gilliamella достигла пика в 36,32% на 1 дпе, затем снизилась до 5,91% на 3 дпе; динамическое равновесие было достигнуто на 7 дпе. Snodgrassella была значительно колонизирована на 1 дпе и была относительно стабильной в микробиоте кишечника. Относительная численность рода Lactobacillus в кишечнике рабочих составляла всего 2,79% на 0 дпе и 5,54% на 1 дпе, а пик относительной численности пришелся на 3 дпе — 48,28%, что указывает на то, что колонизация Lactobacillus произошла после трех дней после появления рабочих. Относительная численность Bifidobacterium в кишечнике только что появившихся рабочих составляла всего 0,58%. Относительная численность Bifidobacterium достигла 12,10% на 3 день и оставалась стабильной с 19 дня. Относительная численность Rhizobiales составляла всего 0,16% в кишечнике только что появившихся рабочих. После достижения 7,24% на 19 дпе относительная численность ризобий оставалась стабильной. Относительная численность Frischella достигла 9,70% на 1 дпе, затем снизилась и стала стабильной. Относительная численность Commensalibacter значительно увеличилась на 3 дпе, а затем также продолжала расти. Относительная численность Apibacter значительно увеличилась на 19 дпе и затем также продолжала увеличиваться. Относительная численность Bartonella значительно увеличилась на 12 дне. Значительное увеличение основной микробиоты сопровождалось значительным уменьшением таких родов, как Acinetobacter, Cyanobacteria, Peptostreptococcaceae, Bacteroidales, Bacteroides и Escherichia-Shigella (рис. 5).
3.4. Преемственность основной микробиоты рабочих
Сравнивая относительное обилие микробиоты кишечника между рабочими 0 dpe и 1 dpe, мы обнаружили, что относительное обилие Acinetobacter значительно снизилось с 18,74% до 0,18% (p = 0,00508), Cyanobacteria снизилось с 6,68% до 1,50% (p = 0,00824), а Xanthomonadaceae снизилось с 1,58% до 0,015% (p = 0,00499). Относительное обилие микробиоты, переносимой рабочими 0 dpe, было значительно снижено по сравнению с рабочими других возрастов, и они были замещены основной микробиотой. Например, Gilliamella увеличилась с 0,0044% до 36,32% (p = 0,00367), Snodgrassella увеличилась с 0,0045% до 9,86% (p = 0,00716), Sphaerochaeta увеличилась с 0,24% до 1,73% (p = 0,0202), а Campylobacter увеличился с 0,11% до 1,08% (p = 0,0306). Кроме того, фришелла увеличилась с 0% до 9,70% (p = 0,00278). Фришелла не была обнаружена в кишечнике вновь появившихся рабочих.
Сравнивая 1 dpe с 3 dpe, мы обнаружили, что это критическое время для колонизации Lactobacillus и Bifidobacterium, поскольку Lactobacillus увеличился с 5,54% до 48,28% (p = 0,00508), а Bifidobacterium увеличился с 0,41% до 12,10% (p = 0,00508). Кроме того, от 1 dpe до 3 dpe также является ключевым временным интервалом колонизации Commensalibacter, которая увеличилась с 0,021% до 2,12% (p = 0,00508). Относительное обилие родов Gilliamella (p = 0,00824), Parabacteroides (p = 0,0202) и Alloprevotella (p = 0,0306) также значительно снизилось.
Сравнивая 7 dpe с 12 dpe, мы обнаружили, что Lactobacillus helsingborgensis снизился с 6,08% до 1,94% (p = 0,00508). Кроме того, Lactobacillus kunkeei увеличился с 0,29% до 2,20% (p = 0,00824), а Bartonella увеличилась с 0,012% до 1,31% (p = 0,0078).
При сравнении 19 dpe с 25 dpe, Bifidobacterium снизились с 8,29% до 2,70% (p = 0,0453), а Commensalibacter увеличились с 10,07% до 3,32% (p = 0,0306). Кроме того, три рода Bacteroides, Escherichia-Shigella и Porphyromonadaceae также значительно уменьшились за этот период (рис. 6).

3.5. Бета-разнообразие бактерий кишечника
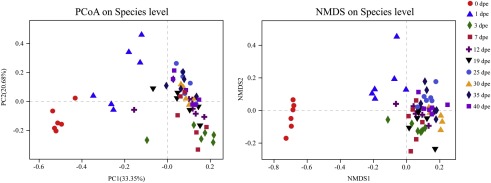
На основе взвешенного расстояния UniFrac или расстояния Брея-Кертиса мы провели анализ главных координат (PCoA) и анализ неметрической многомерной шкалы (NMDS). Кроме того, был проведен анализ сходства (ANOSIM) на основе взвешенного расстояния UniFrac. Результаты показали, что состав микробиоты кишечника коррелировал с возрастом хозяина. Графики PCoA и NMDS показали, что недавно появившиеся рабочие были далеко от других групп, а при сравнении графиков PCoA и NMDS других групп, разделение также было очевидным на 1 дпе, что указывает на то, что относительное обилие основной микробиоты рабочих не было высоким на 1 дпе. Группы 3 дпе и 7 дпе были тесно связаны между собой, а группы 12 дпе и 19 дпе были тесно связаны между собой, несмотря на близкое расстояние между группами на 25 дпе (группа, Am25), 30 дпе, 35 дпе и 40 дпе. В целом, образцы, принадлежащие к каждой группе, были кластеризованы в разных возрастах рабочих. В координатном анализе (PCoA) на долю PC1 пришлось 33,35% общей дисперсии, а на долю PC2 — 20,68%. При анализе неметрической многомерной шкалы (NMDS) стресс = 0,129, что свидетельствовало о надежности группировки и выборки (рис. 7). Кроме того, результаты анализа ANOSIM были следующими: R = 0,4794, P = 0,001. Этот результат дополнительно показывает, что группировка экспериментов была обоснованной.
Обсуждение
В данном исследовании технология высокопроизводительного секвенирования была использована для глубокого секвенирования образцов, взятых в разное время (десять точек отбора). Наши результаты показывают, что состав и структура микрофлоры кишечника рабочих значительно изменяются по мере развития хозяина. Основной микробиом на 0 dpe не был общим с микробиомом на других стадиях, что согласуется с предыдущими исследованиями (Yun et al., 2018). Элайджа Пауэлл и др. обнаружили, что основные микроорганизмы S. alvi, G. apicola и F. perrara значительно колонизируются через социальный контакт (Powell et al., 2014). Поэтому отсутствие основной микрофлоры кишечника у рабочих на 0 дпе может быть объяснено минимальным социальным контактом рабочих. Кроме того, относительная численность Acinetobacter была самой высокой на 0 dpe, что указывает на то, что род Acinetobacter играет важную роль в кишечнике рабочих. Структура флоры кишечника на 0 дпе быстро изменилась на 1 дпе, особенно основные роды, такие как Frischella, Snodgrassella и Gilliamella, что указывает на то, что три основных рода в основном занимают экологическую нишу Acinetobacter. F. perrara вызывает сильную активацию иммунной системы хозяина, и исследования показали, что присутствие F. perrara влияет на иммунитет и гомеостаз кишечника, а предыдущие исследования продемонстрировали, что повышенная численность F. perrara связана с изменением рациона питания и аномальным развитием хозяина (Emery et al., 2017; Maes et al., 2016). Значительные изменения в рационе и среде развития вновь появившихся рабочих, вероятно, сыграли главную роль в объяснении того, почему Frischella впервые колонизировали кишечник рабочих. Относительная численность рода Gilliamella достигает пика на 1 дпе, что свидетельствует о ключевой роли Gilliamella как основного рода в социальном развитии рабочих. Наше предыдущее исследование показало, что численность G. apicola была относительно низкой на 1 дпе (Guo et al., 2015), что может быть объяснено различиями в характере колонизации разными видами кишечных бактерий.
Lactobacillus и Bifidobacterium помогают в усвоении питания и защите пчел. Роды Lactobacillus и Bifidobacterium, выделенные из кишечника пчел, были распылены в улей, на расплод и пыльцу, что вызвало значительное повышение урожая меда (Alberoni et al., 2018). Эти два рода, очевидно, колонизировали между 1 dpe и 3 dpe в данном исследовании, поскольку относительное обилие этих двух видов бактерий значительно увеличилось в этот период (от 1 dpe до 3 dpe). Андерсон и др. исследовали колонизацию Lactobacillus с помощью технологии высокопроизводительного секвенирования. Правило колонизации Lactobacillus было выяснено при различных диетах и социальных контактах, и результаты показали, что процесс колонизации Lactobacillus в задней кишке колебался в зависимости от изменений в питании, улье и социальной среде (Anderson et al., 2016). Кроме того, картина колонизации Lactobacillus в данном исследовании соответствовала данным предыдущих исследований (Anderson et al., 2016; Guo et al., 2015). А именно, предыдущие исследования показали, что различные источники не влияли на режим колонизации Lactobacillus. В период между 1 и 3 дпе численность родов Lactobacillus и Bifidobacterium постоянно увеличивалась. Основные задачи рабочих между 1 и 3 дпе включают очистку клеток, отдых или груминг (Seeley, 1982). Исследования Кенеровой и др. (2017) показали, что Bifidobacterium могут стимулировать хозяина к производству гормонов, которые могут влиять на развитие пчел и ускорять развитие рабочих особей (Kešnerová et al., 2017). Значительно увеличенная относительная численность Bifidobacterium может быть показателем быстрого развития рабочих особей в этот период.
С возрастом пчел их социальные задачи меняются. В период между 4 и 12 годами рабочие пчелы выполняют функции медсестер и носильщиков, передавая белковые выделения младшим и старшим пчелам (Crailsheim, 1992, 1991; Seeley, 1982). Относительная численность Lactobacillus и Bifidobacterium в кишечнике остается стабильной после 4 дпе. Bifidobacterium в кишечнике пчел метаболизируют ряд углеводов, обеспечивая множество питательных веществ и энергии (Bottacini et al., 2012b), а также материал, гарантирующий выполнение социальных задач для рабочих особей.
Кроме того, Lactobacillus и Bifidobacterium имеют множество функций, связанных с разложением углеводов, и было выдвинуто предположение, что группа Lactobacillus, состоящая из Lactobacillus и Bifidobacterium, участвует в переработке нектара и метаболизме углеводов (Butler et al., 2013; Engel et al., 2012). Колонизация этих двух родов способствует перевариванию и усвоению питательных веществ в организме хозяина, что косвенно способствует выполнению социальных задач рабочими.
Lactobacillus kunkeei значительно увеличился с 7 по 12 дпе, и L. kunkeei не имел уникального распределения в кишечной флоре рабочих. L. kunkeei также была обнаружена в других органах рабочих. Например, большое количество L. kunkeei, кислотоустойчивой и осмотолерантной бактерии, обнаружено в хлебе (Anderson et al., 2014). Кроме того, рабочие в этом возрасте в основном занимались переработкой меда (Trumbo et al., 1997). Таким образом, мы предположили, что L. kunkeei является группой ключевых бактерий в микробиоте кишечника рабочих для производства меда, а функция сопротивления рабочих к кислотным и осмотолерантным повреждениям, вызванным производством меда, формировалась с 7 по 12 дпе.
Обилие Bifidobacterium было значительно снижено между 9 и 30 дпе. В это время рабочие покидают ульи и сосредотачиваются на поиске пыльцы, нектара, прополиса и воды (Calderone, 1998; Robinson, 1992). Это может быть одной из причин снижения относительной численности рода Bifidobacterium, как сообщается в исследованиях Anderson et al. (2018), которые обнаружили, что относительная численность Bifidobacterium значительно снижается с возрастом рабочих (Anderson et al., 2018), что аналогично результатам, полученным в текущем исследовании. Кроме того, F. perrara также значительно снижается из-за изменений в окружающей среде и рационе, когда рабочие покидают улей для выполнения задач по сбору (Martinson et al., 2012). Это наблюдение показывает, что изменение относительной численности F. perrara соответствует изменению численности Bifidobacterium в этот период. Однако род Bifidobacterium является бактериальным индикатором для оценки возраста рабочих (Anderson et al., 2018). Полученные здесь результаты указывают на потенциал F. perrara в качестве индикатора для оценки возраста, но это предположение требует дальнейшего изучения в будущем.
В целом, здоровье медоносных пчел, как важного экономического насекомого, вызывает озабоченность. В данном исследовании было проведено высокопроизводительное секвенирование гена 16S рРНК для микрофлоры кишечника пчел на различных стадиях развития. Мы обнаружили, что структура микрофлоры кишечника пчел значительно изменялась в зависимости от возрастного развития. Рабочие, достигшие 0 дпе, не имели в кишечнике основной кишечной флоры, а ключевые моменты для колонизации основной кишечной флоры наступали на 1-3 дпе. Кроме того, мы выяснили правила динамической микробной сукцессии в кишечнике пчел. Изменение микрофлоры кишечника значительно коррелировало с увеличением суточного возраста. Это исследование не только углубляет наше понимание паттерна колонизации основной микрофлоры кишечника у рабочих особей, но и предоставляет полезную информацию для углубленного изучения процессов колонизации микрофлоры кишечника пчел.
Приложение А. Дополнительные данные
Ниже приведены дополнительные данные к этой статье:
Ссылки
1.Alberoni et al., 2018 D. Alberoni, L. Baffoni, F. Gaggìa, P. Ryan, K. Murphy, P.R. Ross, C. Stanton, D. Di Gioia
Impact of beneficial bacteria supplementation on the gut microbiota, colony development and productivity of Apis mellifera L. Benef Microbes, 9 (2018), pp. 269-278
View article CrossRef View in Scopus Google Scholar
2.Anderson et al., 2014 K.E. Anderson, M.J. Carroll, T. Sheehan, M.C. Lanan, B.M. Mott, P. Maes, V. Corby-Harris
Hive-stored pollen of honey bees: many lines of evidence are consistent with pollen preservation, not nutrient conversion Mol. Ecol., 23 (2014), pp. 5904-5917 View PDF This article is free to access.
CrossRef View in Scopus Google Scholar
3.Anderson et al., 2018 K.E. Anderson, V.A. Ricigliano, B.M. Mott, C.C. Duan, A.S. Floyd, P. Maes The queen’s gut refines with age: longevity phenotypes in a social insect model Microbiome, 6 (2018), p. 108 View PDF This article is free to access.
View in Scopus Google Scholar
4.Anderson et al., 2016 K.E. Anderson, P.A.P. Rodrigues, B.M. Mott, P. Maes, V. Corby-Harris Ecological succession in the honey bee gut: shift in Lactobacillus strain dominance during early adult development Mol. Ecol., 71 (2016), pp. 1008-1019
View article CrossRef View in Scopu sGoogle Scholar
5.Babendreier et al., 2007 D. Babendreier, D. Joller, J. Romeis, F. Bigler, F. Widmer Bacterial community structures in honeybee intestines and their response to two insecticidal proteins FEMS Microbiol. Ecol., 59 (2007), pp. 600-610
View PDF CrossRef View in Scopus Google Scholar
6.Bottacini et al., 2012a F. Bottacini, C. Milani, F. Turroni, nB Sã, E. Foroni, S. Duranti, F. Serafini, A. Viappiani, F. Strati, A. Ferrarini Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect gut PLoS One, 7 (2012), p. e44229
View PDF CrossRef View in Scopus Google Scholar
7.Bottacini et al., 2012b F. Bottacini, C. Milani, F. Turroni, B. Sánchez, E. Foroni, S. Duranti, F. Serafini, A. Viappiani, F. Strati, A. Ferrarini, M. Delledonne, B. Henrissat, P. Coutinho, G.F. Fitzgerald, A. Margolles, D. van Sinderen, M. Ventura Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect gut PLoS One, 7 (2012) e44229-e44229
Google Scholar
8.Butler et al., 2013 È. Butler, M. Alsterfjord, T.C. Olofsson, C. Karlsson, J. Malmström, A. Vásquez Proteins of novel lactic acid bacteria from Apis mellifera mellifera: an insight into the production of known extra-cellular proteins during microbial stress BMC Microbiol., 13 (2013), p. 235
View PDF This article is free to access. View in Scopus Google Scholar
9.Calderone, 1998 N.W. Calderone Proximate mechanisms of age polyethism in the honey bee, Apis mellifera L Apidologie, 29 (1998), pp. 127-158 View PDF CrossRef View in Scopus Google Scholar
- 10.Cameron et al., 2011 S.A. Cameron, J.D. Lozier, J.P. Strange, J.B. Koch, N. Cordes, L.F. Solter, T.L. Griswold Patterns of widespread decline in North American bumble bees Proc. Natl. Acad. Sci. U. S. A., 108 (2011), p. 662
- View PDF CrossRef View in Scopus Google Scholar
11.Colin et al., 2019 T. Colin, W.G. Meikle, A.M. Paten, A.B. Barron Long-term dynamics of honey bee colonies following exposure to chemical stress Sci. Total Environ., 677 (2019), pp. 660-670
View PDF View article View in Scopus Google Scholar
12.Crailsheim, 1992 K. Crailsheim The flow of jelly within a honeybee colony J. Comp. Physiol. B, 162 (1992), pp. 681-689
View in Scopus Google Scholar
13.Crailsheim, 1991 K. Crailsheim Interadult feeding of jelly in honeybee (Apis mellifera L.) colonies J. Comp. Physiol. B, 161 (1991), pp. 55-60 View in Scopus Google Scholar
14.Emery et al., 2017 O. Emery, K. Schmidt, P. Engel Immune system stimulation by the gut symbiont Frischella perrara in the honey bee (Apis mellifera) Mol. Ecol., 26 (2017), pp. 2576-2590
View article CrossRef View in Scopus Google Scholar
15. Engel et al., 2012 P. Engel, V.G. Martinson, N.A. Moran Functional diversity within the simple gut microbiota of the honey bee Proc. Natl. Acad. Sci. U. S. A., 109 (2012), pp. 11002-11007
View PDF CrossRef View in Scopus Google Scholar
16.Guo et al., 2015 J. Guo, J. Wu, Y. Chen, J.D. Evans, R. Dai, W. Luo, J. Li Characterization of gut bacteria at different developmental stages of Asian honey bees, Apis cerana J. Invertebr. Pathol., 127 (2015), pp. 110-114
View PDF View article View in Scopus Google Scholar
17.Hauke and Paul, 2011 K. Hauke, S.H. Paul Socially transmitted gut microbiota protect bumble bees against an intestinal parasite Proc. Natl. Acad. Sci. U. S. A., 108 (2011), pp. 19288-19292
Google Scholar
18.Huang et al., 2018 S.K. Huang, K.T. Ye, W.F. Huang, B.H. Ying, X. Su, L.H. Lin, J.H. Li, Y.P. Chen, J.L. Li, X.L. Bao, J.Z. Hu Influence of feeding type and Nosema ceranae infection on the gut microbiota of Apis cerana workers mSystems, 3 (2018), pp. e00177-118
View PDF This article is free to access. View in Scopus Google Scholar
19.Jeyaprakash et al., 2003 A. Jeyaprakash, M.A. Hoy, M.H. Allsopp Bacterial diversity in worker adults of Apis mellifera capensis and Apis mellifera scutellata (Insecta: hymenoptera) assessed using 16S rRNA sequences J. Invertebr. Pathol., 84 (2003), pp. 96-103
View PDF View article View in Scopus Google Scholar
20.Jones et al., 2017 J.C. Jones, C. Fruciano, F. Hildebrand, H. Al Toufalilia, N.J. Balfour, P. Bork, P. Engel, F.L. Ratnieks, W.O. Hughes Gut microbiota composition is associated with environmental landscape in honey bees Ecol. Evol., 8 (2017), pp. 441-451
Google Scholar
21.Kakumanu et al., 2016 M.L. Kakumanu, A.M. Reeves, T.D. Anderson, R.R. Rodrigues, M.A. Williams Honey bee gut microbiome is altered by in-hive pesticide exposures Front. Microbiol., 7 (2016) 1255-1255
Google Scholar
22. Kešnerová et al., 2017 L. Kešnerová, R.A.T. Mars, K.M. Ellegaard, M. Troilo, U. Sauer, P. Engel Disentangling metabolic functions of bacteria in the honey bee gut PLoS Biol., 15 (2017) e2003467-e2003467
Google Scholar
23.Kwong and Moran, 2013 W.K. Kwong, N.A. Moran Cultivation and characterization of the gut symbionts of honey bees and bumble bees: description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a membe Int. J. Syst. Evol. Microbiol., 63 (2013), pp. 2008-2018
View article CrossRef View in Scopus Google Scholar
24. Kwong and Moran, 2016 W.K. Kwong, N.A. Moran Gut microbial communities of social bees Nat. Rev. Microbiol., 14 (2016), p. 374
View article CrossRef View in Scopus Google Scholar
25.Langowska et al., 2017 A. Langowska, M. Zawilak, T.H. Sparks, A. Glazaczow, P.W. Tomkins, P. Tryjanowski Long-term effect of temperature on honey yield and honeybee phenology Int. J. Biometeorol., 61 (2017), pp. 1125-1132
View PDF This article is free to access. CrossRef View in Scopus Google Scholar
26.Liu et al., 2011 P.D. Liu, J. Wu, H.Y. Li, S.W. Lin Economic values of bee pollination to China’s agriculture Sci. Agric. Sin. (in chinese), 44 (2011), pp. 5117-5123 Google Scholar
27.Maes et al., 2016 P.W. Maes, P.A. Rodrigues, R. Oliver, B.M. Mott, K.E. Anderson Diet-related gut bacterial dysbiosis correlates with impaired development, increased mortality and Nosema disease in the honeybee (Apis mellifera) Mol. Ecol., 25 (2016), pp. 5439-5450
View article CrossRef View in Scopus Google Scholar
28.Martinson et al., 2011 V.G. Martinson, B.N. Danforth, R.L. Minckley, R. Olav, T. Salim, N.A. Moran A simple and distinctive microbiota associated with honey bees and bumble bees Mol. Ecol., 20 (2011), pp. 619-628
View in Scopus Google Scholar
29.Martinson et al., 2012 V.G. Martinson, J. Moy, N.A. Moran Establishment of characteristic gut bacteria during development of the honeybee worker Appl. Environ. Microbiol., 78 (2012), pp. 2830-2840
View PDF This article is free to access. View in Scopus Google Scholar
30.Moran et al., 2003 N.A. Moran, G.R. Plague, J.P. Sandström, J.L. Wilcox A genomic perspective on nutrient provisioning by bacterial symbionts of insects Appl. Environ. Microbiol., 100 (2003), pp. 14543-14548
View PDF View in Scopus Google Scholar
31.Motta et al., 2018 E.V.S. Motta, K. Raymann, N.A. Moran Glyphosate perturbs the gut microbiota of honey bees Proc. Natl. Acad. Sci. U. S. A., 115 (2018), p. 10305
View PDF CrossRef View in Scopus Google Scholar
32.Owen, 2017 R. Owen Role of human action in the spread of honey bee (Hymenoptera: apidae) pathogens J. Econ. Entomol., 110 (2017), pp. 797-801
View article CrossRef View in Scopus Google Scholar
33.Philipp et al., 2013 E. Philipp, W.K. Kwong, N.A. Moran Frischella perrara gen. nov., sp. nov., a gammaproteobacterium isolated from the gut of the honeybee, Apis mellifera Int. J. Syst. Evol. Microbiol., 63 (2013), pp. 3646-3651
Google Scholar
34. Powell et al., 2014 J.E. Powell, V.G. Martinson, K. Urban-Mead, N.A. Moran Routes of acquisition of the gut microbiota of the honey bee Apis mellifera Appl. Environ. Microb., 80 (2014), pp. 7378-7387
View PDF This article is free to access. View in Scopus Google Scholar
35.Raymann et al., 2017 K. Raymann, Z. Shaffer, N.A. Moran Antibiotic exposure pertubs the gut micxrobiota and elevates mortality in honeybees PLoS Biol., 15 (2017), p. e2001861
View PDF CrossRef View in Scopus Google Scholar
36.Robinson, 1992 G.E. Robinson Regulation of division of labor in insect societies Annu. Rev. Entomol., 37 (1992), pp. 637-665
View article CrossRef View in Scopus Google Scholar
37.Ross, 2012 R.P. Ross Composition of the early intestinal microbiota:knowledge, knowledge gaps and the use of high-throughput sequencing to address these gaps Gut Microbes, 3 (2012), pp. 203-220 View in Scopus Google Scholar
38.Sabree et al., 2012 Z.L. Sabree, A.K. Hansen, N.A. Moran Independent studies using deep sequencing resolve the same set of core bacterial species dominating gut communities of honey bees PLoS One, 7 (2012), p. e41250
View PDF CrossRef View in Scopus Google Scholar
39.Seeley, 1982 T.D. Seeley Adaptive significance of the age polyethism schedule in honeybee colonies Behav. Ecol. Sociobio., 11 (1982), pp. 287-293 View in Scopus Google Scholar
40.Southwick and Southwick, 1992 E.E. Southwick, L. Southwick Estimating the economic value of honey bees (Hymenoptera: apidae) as agricultural pollinators in the United States J. Econ. Entomol., 85 (1992), pp. 621-633
View article CrossRef View in Scopus Google Scholar
41.Stokstad, 2007 E. Stokstad The case of the empty hives Science, 316 (2007), pp. 970-972
View article CrossRef View in Scopus Google Scholar
42.Trumbo et al., 1997 S.T. Trumbo, Z.-Y. Huang, G.E. Robinson Division of labor between undertaker specialists and other middle-aged workers in honey bee colonies Behav. Ecol. Sociobio., 41 (1997), pp. 151-163
View in Scopus Google Scholar
43.Vanessa et al., 2014 C.H. Vanessa, L.A. Snyder, M.R. Schwan, M. Patrick, Q.S. McFrederick, K.E. Anderson Origin and effect of Alpha 2.2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. Nov Appl. Environ. Microbiol., 80 (2014), pp. 7460-7472
Google Scholar
44.Yun et al., 2018 J.H. Yun, M.J. Jung, P.S. Kim, J.W. Bae Social status shapes the bacterial and fungal gut communities of the honey bee Sci. Rep., 8 (2018), p. 2019
View PDF This article is free to access. View in Scopus Google Scholar
45.Zhang et al., 2018 F. Zhang, X.X. Sun, X.C. Zhang, S. Zhang, J. Lu, Y.M. Xia, Y.H. Huang, X.J. Wang The interactions between gut microbiota and entomopathogenic fungi: a potential approach for biological control of Blattella germanica (L.) Pest Manag. Sci., 74 (2018), pp. 438-447
View article CrossRef View in Scopus Google Scholar
Добавить комментарий